Species of Interest






Lines of Research
The MYB (myeloblastosis) family is one of the biggest TF (super)families ever described in plants (Riechmann et al., 2000). They were first identified in mammals (reviewed by Oh & Reddy, 1999) but are present in other animal and also fungal genomes (Du et al., 2015). Our interest in this family of transcriptional regulators comes from the impact of some of its members in the regulation of plant secondary metabolism. However, on the grounds that this family became largely expanded through the course of plant evolution, MYB proteins actually regulate a plethora of other processes, including development, hormone signaling and the coordination of responses to cope with stress. In fruit and horticulture crops, they are responsible for many desired consumer traits such as color, taste, flavor and texture (reviewed by Allan and Espley, 2018).
In plants different MYB subfamilies evolved, being identified by the number of imperfect repeats (named R) present in the N-terminal region. These repeats normally constitute the main DNA binding domains, normally 100–160 residues in length. The R2R3 subfamily is the most abundant in plants (more than 120 members in Arabidopsis, tomato or grape). Here, each repeat adopts a helix-helix-turn-helix structure that altogether interact with DNA regulatory elements. Residues in the DNA-binding domain may also give specificity for co-factor interaction and for gene target selection, while different motifs found in the C-terminal region can establish protein-protein interactions with other regulators or with components of the eukaryotic transcriptional machinery (Figure 1).
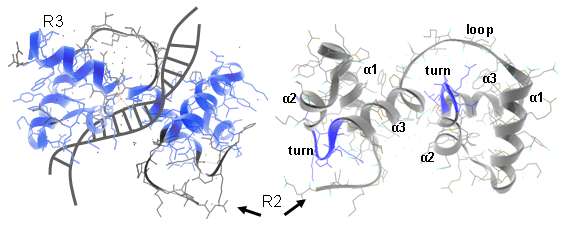
Figure 1. Ribbon diagram of the rat c-MYB R2R3 domain and its DNA-interaction interface. Left panels show R2R3 domain in the opposite orientation. α helices are numbered (Matus et al., 2008).
We attempt to characterize the MYB family in different plant species by combining genome-wide studies and in planta functional characterizations. Our main attention is in the regulation of phenylpropanoid and isoprenoid metabolisms, in part responsible for the beneficial attributes of the so-called ‘functional foods’ (those containing various factors enhancing health). Among the many phenylpropanoid compounds that influence the quality of fruits and their derived products (such as grapes and wines), some of the most important are the flavonoid group (i.e. anthocyanins, flavonols and tannins) and stilbenes. These compounds influence aromas, color, astringency and taste, and provide many beneficial attributes towards human health when consumed (nutraceutical properties such as anti-cancer, antioxidant, UV screens, anti-virus, anti-inflammatory, wound healing, antibacterial, etc.). For example, in humans, the intake of resveratrol (i.e. the most abundant stilbene found in many fruits) lowers the risks of cardiovascular disease and its antioxidant, anti-aging and anti-angiogenesic properties are well recognized and have been subject of numerous research endeavors.
We have originally selected grape, tomato and Arabidopsis as the main subject species to characterize the R2R3-MYB families due to their importance as economical or plant model species. On grapevine, the R2R3-MYB family was initially assessed in the cv. ‘Pinot Noir’ genome by Matus et al. (2008), showing it possessed more than 100 genes. Interestingly, wine quality clades associated to the flavonoid pathway were found to be expanded, many of which could have been fixed during the course of domestication. Recently we adopted a systems biology approach to re-update the family gathering more than 130 members (Figure 2; Wong et al., 2016).
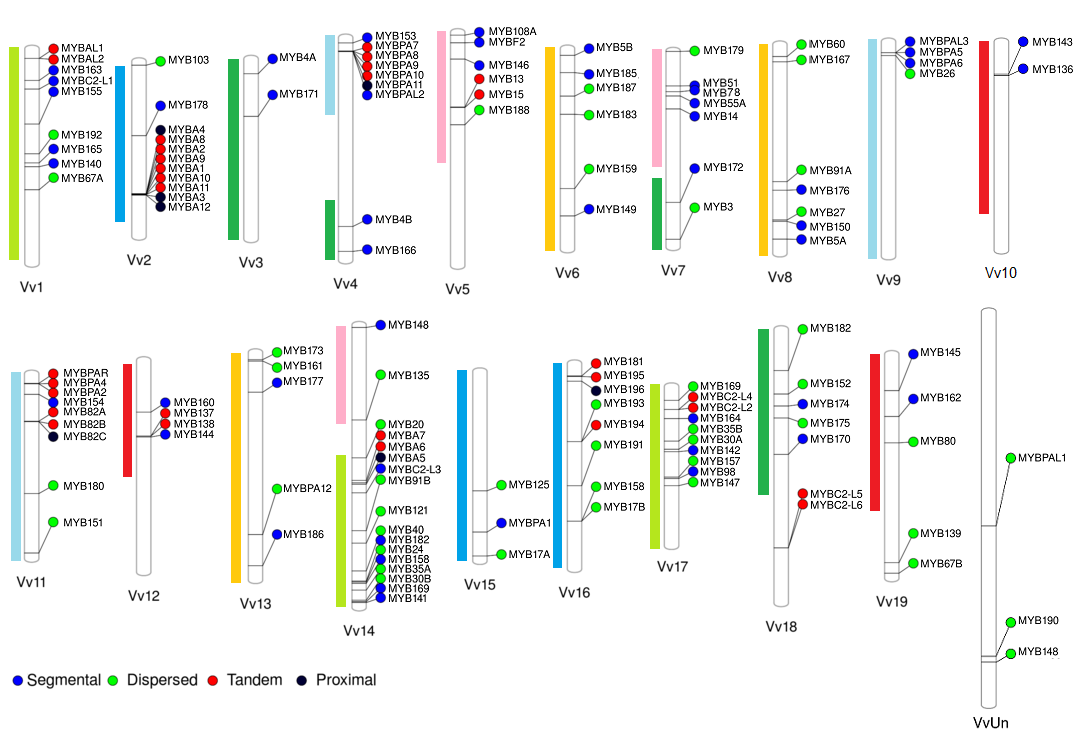
Figure 2. Distribution of R2R3-MYB genes in the cv. ‘Pinot Noir’ genome (for more details, see Wong et al., 2016). Gene names were adopted according to the Super Nomenclature Committee.
For additional resources on plant MYB gene families go here.
We explored the capacity of co-expression networks to provide a wide overview of gene function and expression behavior of MYB genes in this species. The objective of this first glimpse into network analysis was to find the biological processes regulated by grapevine R2R3-MYBs through the combination of gene expression data, phylogenetic information, DNA-binding motif discovery and experimental evidence. MYB gene-centered co-expression networks were constructed from publicly available microarray data to predict the function and potential downstream targets of the complete grapevine R2R3-MYB family. The top 100-ranked co-expressed genes for each grape gene were selected and inspected, showing that many MYB genes were associated not only to the regulation of phenylpropanoids (as previously hypothesized) but also to the isoprenoid pathway, responsible for the synthesis of carotenoids, tocopherols and terpene volatile compounds.
In the forthcoming years, we intend to continue characterizing this family in grapevine by adding novel layers of genome-wide data and by updating co-expression networks after the incorporation of RNA-seq datasets. We intend to select MYB homologues and characterize their function in other species such as tomato and Arabidopsis. Altogether with these activities, we intend to determine the gene regulatory networks for each R2R3-MYB.
Line 2a: Identifying novel regulators and enzymes from secondary metabolic pathways
Plants produce a large variety of natural products that are important for species interactions, including the communication of plants with other organisms. When consumed by humans, many of these plant-derived secondary metabolites engender dietary benefits and play important roles in disease prevention, leading to a longer and healthier life. Understanding how these compounds are produced and spatially and temporally-regulated has an enormous potential for the agro-industry sector, impacting molecular breeding programs and favoring the development of strategies for engineering healthier foods. As plant-derived compounds have the potential to be produced on an industrial scale, this knowledge can also be transferred to the flavor and pharmaceutical industries.
Among our research interests, we intend to combine transcriptomic and metabolomic data to identify unknown secondary metabolic enzymes and their transcriptional regulators in diverse plant species, which may be important for drug discovery and improvement of functional foods. This systems biology approach will be further complemented with experimental data generated from in vitro and in planta gene characterizations.
The most extensive progress in the identification of transcriptional regulators of secondary metabolism comes from the study in plant model species (e.g. maize, petunia and Arabidopsis) of the control of flavonoid metabolism. These studies have grounded the basis of a eudicot-conserved model for the regulation of this metabolic pathway, exerted by a combinatory interaction between R2R3-MYB activators, R3 and R2R3-MYB repressors, beta helix-loop-helix (bHLH/MYC) co-regulators and trypthophan-aspartic acid repeat (WDR) proteins that form a dynamic complex (MBW; Albert et al., 2015). To date, several other families of transcription factors (TFs) have been implicated in the control of these pathways. Such is the case of WRKY and NAC family members recently characterized in fruit crop species (Amato et al., 2017; Zhou et al., 2015). In grapevine, several genes coding for regulators of the phenylpropanoid pathway have been isolated and characterized in the last decade (Figure 2.1). In fact, this species is an interesting model for examining the regulation of metabolism because of the expansion and diversification of genetic factors controlling the synthesis of secondary metabolites.

Figure 2.1. Multi-layered transcriptional regulation of secondary metabolism exerted by different transcription factor (TF) families. Berry development is described in resemblance to a clock, with the fruit set–to–post harvest-withering (PHW) stages shown in anticlockwise direction. Veraison corresponds to the onset of ripening. The secondary metabolic pathways controlled by these regulators are shown on top of each group of TFs (Adapted from Matus, 2016). UVR: UV-B responsive. (+): WRKYs may activate stilbene synthases alone or in combination with MYB14 (Vannozzi et al., 2018).
Transcriptomics and metabolomics data typically provide a global ‘snapshot’ of gene expression and metabolite dynamics in various biological contexts. Together, they constitute layers of biological complexity. For many omics data, molecular associations between molecules are often represented as networks containing a large collection of nodes and edges. Edges connecting nodes represent associations often inferred from correlational and informational theoretic measures. Gene co-expression network (GCN) analyses are based on the “guilt by association” principle where genes (nodes) involved in similar or related processes may share similar gene expression dynamics (edges) across a wide range of experiments. GCN analyses have become a popular and powerful tool for inferring gene function using large transcriptomics datasets with much success. An increasing number of studies have used gene co-expression networks to find common pathways and putative targets for transcription factors (TFs) related to secondary metabolism. As an example, we recently constructed a composite network for stilbene synthase regulation with the aim of illustrating different approaches of data integration for network analysis in grapevine (Wong and Matus, 2017). Using publicly available berry-specific RNA-Seq data, we overlapped gene co-expression networks (GCNs) with the presence of promoter cis-binding elements (CREs), microRNAs and long non-coding RNAs. As a result, we inferred a systems-level STS regulatory network from the context of berry ripening (Figure 2.2).
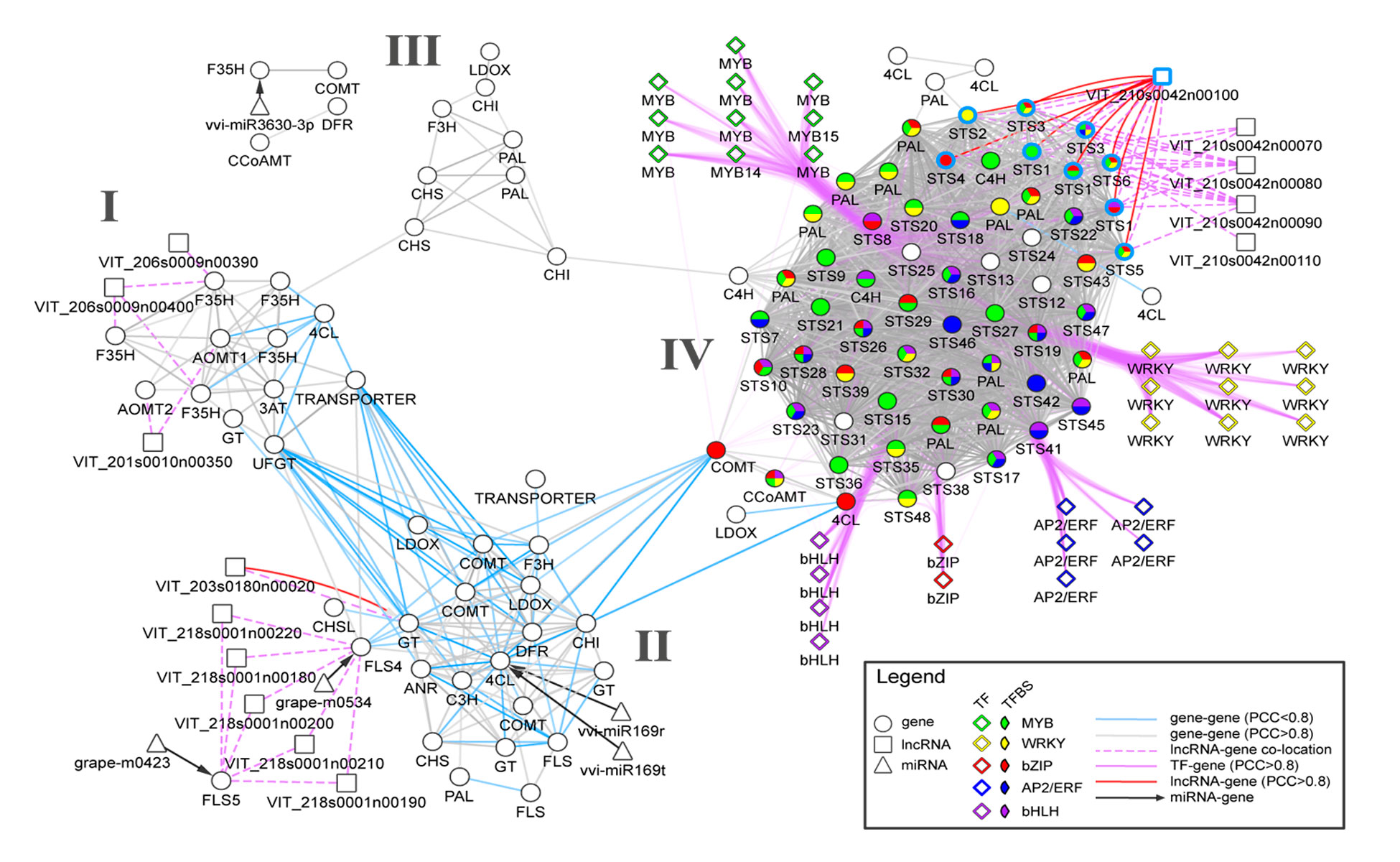
Figure 2.2. An integrated network for phenylpropanoid regulation in the ripening grape berry. Taken from Wong and Matus, 2017. Frontiers in Plant Science.
In the re-analysis of the grape R2R3-MYB family (Wong et al., 2016) we suggested that many of its members participated in secondary metabolic processes and that this regulation was beyond the phenylpropanoid pathway. Representation analysis of the MapMan BIN terms for their top100 co-expressed genes showed that several MYB genes were highly associated with isoprenoids. In fact, recent studies have shown that MYBs can directly regulate sesquiterpene-lactone or carotenoid biosynthesis in Artemisia and Mimulus, respectively (Matías‐Hernández et al., 2017; Sagawa et al., 2016). Taking all this in mind, in the forthcoming years we expect to integrate genomic, transcriptomic and metabolite networks in different species holding valuable secondary compounds to fully characterize their metabolic pathways and understand their regulation.
Line 2b: Inter-relation between different secondary metabolic pathways in plants.
Many secondary metabolites provide pigment traits in plants. Most of them accumulate as the result of the activity of the flavonoid (anthocyanin) and isoprenoid (carotenoid) pathways. Partly, their presence in flowers and fruits has allowed plant co-evolution with insects and seed dispersers. Additionally, they also filter harmful excessive radiation and provide accessory photosynthetic capacity. To date, the interrelation between these distinct pathways is unclear: up to what point may they compete for primary intermediates? are they regulated by similar TFs? or is there some sort of feedback communication between them? With the purpose of testing the balancing relations between branches of secondary metabolic pathways in crop plants, we are examining the effect of pigment depletion or emergence by addressing a) grapevine color somatic variants (e.g. Ferreira et al., 2019; Santibáñez et al., 2019), b) a grape naturally-occurring and infrequent berry skin color variegation (unpublished data) and c) the effects of overproducing anthocyanin pigments in carotenoid-rich fruits such as tomato. While the emergence of anthocyanin pigments is accompanied with parallel branch pathways such as stilbene accumulation, the lack of pigments is generally associated with the increased accumulation of metabolites of different chemical structure (i.e. flavonoids and some isoprenoids). We are currently performing transcriptome and metabolome integrated analyses to determine the causes and consequence of pigment depletion/emergence. Our results suggest controlled balances between distant pathways and which may imply a tight regulation of a transcription factor for such balances.
Based on the results generated in all of our related activities, we have created a publicly available web-based platform with several Apps. The Plantae Visualization platform provides a range of visualization and analysis tools drawn from a range of big data sources in several plant species and microalgae.
Referencing PlantaeViz: Santiago, A., Orduña, L., Fernández, J. D., Vidal, Á., De Martín-Agirre, I., Lisón, P., Vidal, E. A., Navarro-Payá, D., & Matus, J. T. (2024). The Plantae Visualization Platform: A comprehensive web-based tool for the integration, visualization, and analysis of omic data across plant and related species. bioRxiv. https://doi.org/10.1101/2024.12.19.629382.
The Gene Cards app provides a gene-centered view of literature-based functional associations, amongst other information, found in the reference gene catalogue (Navarro-Payá et al., 2022a; https://integrape.eu/resources/). Gene Cards also provides tissue specific boxplots showing tissue-specific gene expressions (log(FPKM + 1)) across more than 2,700 curated RNA-seq SRA studies.
The Expression Atlases (EX-ATLAS) app allows to navigate through different transcriptomic datasets such as the cv. ‘Corvina’ organ atlas, the cv. ‘Pinot’ flower atlas or a berry development time-course atlas, presented as heatmaps or plots.
Tissue specific aggregate gene co-expression networks (GCNs) using more than 2,700 RNA-seq SRA studies can be visualized and analyzed using the different functionalities within the AggGCNs app.
The EXHARA app allows to view the same expression data used in the networks in a heatmap form.
Conversions app provides up-to-date equivalences between current and past PN40024 reference genome annotation versions.
DAP-Browse app (Navarro et al., 2022b) brings together DNA affinity purification sequencing (DAP-seq) data produced by the community on different grapevine transcription factors. It consists of a JBrowse program tailored for DAP-seq data track visualization in the context of the PN40024 grapevine reference genome.
The TransMetaDB app (Savoi et al., 2022) can be used to explore the correlation between the expression of a list of genes of interest and different metabolites. For those correctly annotated experiments, there is also the option to merge different studies as a sort of meta-analysis, giving rise to more robust correlation analyses.

The Plantae Visualization platforms in several other species, including arabidopsis (AraViz), mulberry (MulViz), tomato (TomViz), cannabis (CannaViz), among others.
4a. Plant-plant Communication
Plants usually live in groups, conforming communities of individuals of the same or different species. The ecological interaction in this groups usually involve the generation, sensing and transduction of signals that, based on their nature, can be classified as physical (e.g., light) and chemical (e.g., exudates and volatile organic compounds, VOCs). When a plant produces signals that are recognized as information by another plant, they are referred to as emitter and receiver plants, respectively.
Plants emit VOCs from leaves, flowers and fruits into the atmosphere and from roots into the soil. Amongst the vast array of plant compounds secreted to the environment with a potential role as signals in interplant communication, ENIGmA is focused in VOCs produced by leaves that influence light and defense responses.
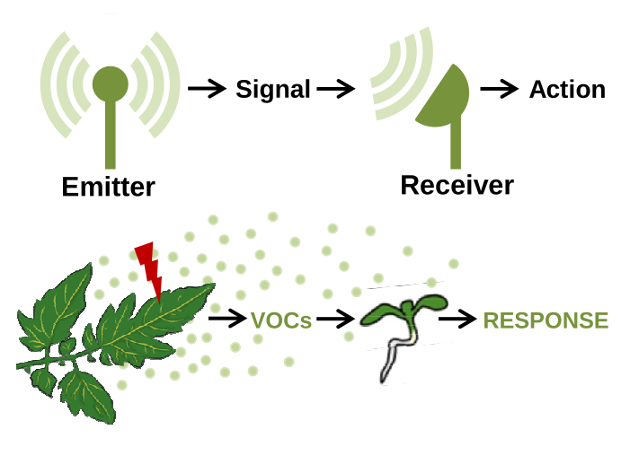
Figure 1. Basic steps in plant-to-plant communication. Messages (signals) produced by emitter plants are transmitted to distant receiver plants, that interpret and respond to the signal.
Plant VOCs are mainly represented by secondary metabolites such as terpenoids (also known as isoprenoids), fatty acid derivatives, benzenoids and phenylpropanoids (Ueda et al., 2012). Some VOCs are synthesized constitutively, including, for instance, a range of VOCs produced by flowers to attract pollinators. Most often, however, VOC levels change in response to environmental signals (Cortes et al., 2016; Kesser & Baldwin, 2001).
While major efforts have been made to investigate the production of VOCs by emitter plants and to unveil the nature of the signals, the processes taking place in the receiver plants have received much less attention. How light signals are perceived and integrated is relatively well understood, but we know virtually nothing of how non-shaded plants respond to the VOCs released by emitter plants exposed lower quality light. How these VOC signals are integrated with others informing of additional environmental challenges (e.g. microbial infection) also remains a mystery.
Our project ENIGmA (funded by GVA Prometeo Program) aims to fill this gap by focusing on the analyses of receiver plants exposed to VOC signals released after shade and pathogen attack. Visit ENIGmA own website here.
In this project, our group will be focused on characterizing the receiver plant’s transcriptomic responses to different VOCs in order to identify putative regulators and components of the sensing and signal transduction mechanisms, as well as derived responses. By comparing the transcriptome of tomato and A. thaliana seedlings exposed to VOCs from emitter plants of the same or a different species, we will further unveil the extent of intraspecific and interspecific information encrypted in VOC signals. Specifically, our tasks include:
– Time-course mRNA sequencing. Tomato and A. thaliana seedlings will be exposed to VOCs produced from the specific combinations of treatments of the 3 emitter species. Total RNA from receiver seedlings will be extracted at different time points of VOC exposure and sequenced with Illumina.
– Differential gene expression analysis. High quality reads will be mapped on each reference genome, identifying co-expression modules that reflect the complete expression trajectories when comparing treatments.
– Selection of candidates for DAP-seq. The best gene candidates encoding transcription factors (Tfs) that appear in the modules of early responses will be selected and used to determine their binding sites by DNA-affinity purification followed by sequencing (DAP-seq).
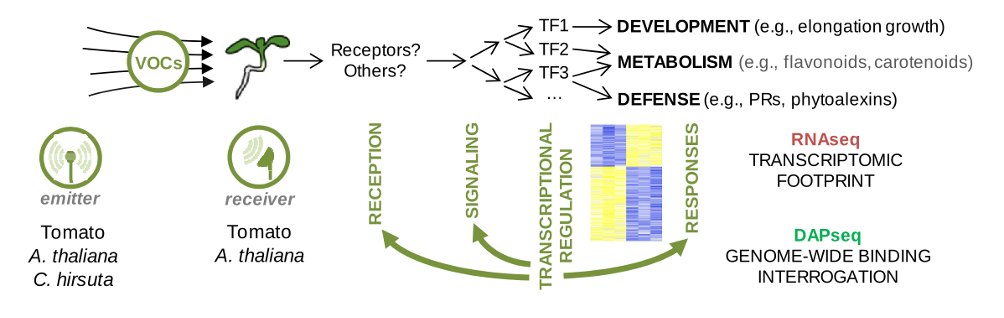
Figure 2. RNA from receiver seedlings will be extracted after the exposure to VOCs produced by emitter plants. After RNA-seq, computational methods will classify early and late transcriptomic changes. DAP-seq will be later conducted to identify early-changing transcription factors (TFs) that may be orchestrating downstream responses. Despite reception and signaling normally precede transcriptomic changes, feedback loops might be revealed by analyzing RNA-seq/DAP-seq datasets.
4b. Plant-insect Communication
Vibroacoustic (VA) signals combine airborne sound and substrate-borne vibrations. Although sounds and vibrations are among the most widespread means of animal communication (Wilkins et al. 2013), for which they fulfill key roles as mating, defence, or social coordination (e.g., Ryan & Cummings 2013; Suzuki 2011; Barbero et al. 2009), a growing body of evidence documents a wide variety of plant responses to VAs (e.g., De Luca & Vallejo-Marin 2013; Appel & Cocroft 2014; Mishra et al 2016). These responses can greatly impact several aspects of plant biology and ecology. Among these, changes in the growth rate of germination (e.g., Jung et al. 2018, Chowdhury et al. 2014), thigmo-morphogenetic responses (Telewski, 2006, 2021), increased photosynthetic ability (Meng et al. 2012), modifications in gene expression (Jeong et al 2008) or phytohormone production (Bochu et al. 2004) were detected in different plant species in response to VA stimulations (Gagliano et al. 2012; Gagliano 2013; Joshi et al. 2019).
VAs could be significant for plants in perceiving and reacting to signals from the surrounding environment or could have emerged as adaptations of the plant in its function as a biotremological communication channel (Oberst et al. 2019). In plant-plant communication, a plant facing drought can produce VAs to signal an incoming threat to neighbouring plants that start closing their stomata in response (Falik et al. 2011). VAs can also be used in plant-insect communication acting as alarming or benevolent signals. Sounds produced by chewing herbivores were observed to promote plant defences against herbivores (Appel & Cocroft 2014). In contrast, substrate vibration (sonication) of buzzing bees can enhance plant’s pollen release, thus increasing pollination (De Luca & Vallejo-Marin 2013). Similarly, bat-pollinated plants have evolved morphological structures to increase the attraction of specific mammal pollinators by exploiting their echolocation systems (Schöner et al. 2016). Up to now the complex communications between flowering plants and pollinators had been extensively studied primarily by assessing the production and perception of visual and olfactory cues (Kantsa et al 2018). To the best of our knowledge, there is only one preliminary work correlating an improvement in plant rewards to pollinator VA signalling. Studying the evening primrose (Oenothera drummondii), Veits and colleagues (2019) found that a rapid sweetness enrichment in nectar occurred after the plants were treated with pollinator VA stimuli. Still, this study did not perform a detailed quantification of sugar types throughout different time-points, neither inspected underlying molecular mechanisms nor tested the plant reproductive success experimentally.
Not only the ultimate functions of VAs in plant biology are poorly understood, but there is a gap in the knowledge also in the proximate mechanisms of VA productions, perception, transduction and transmission.
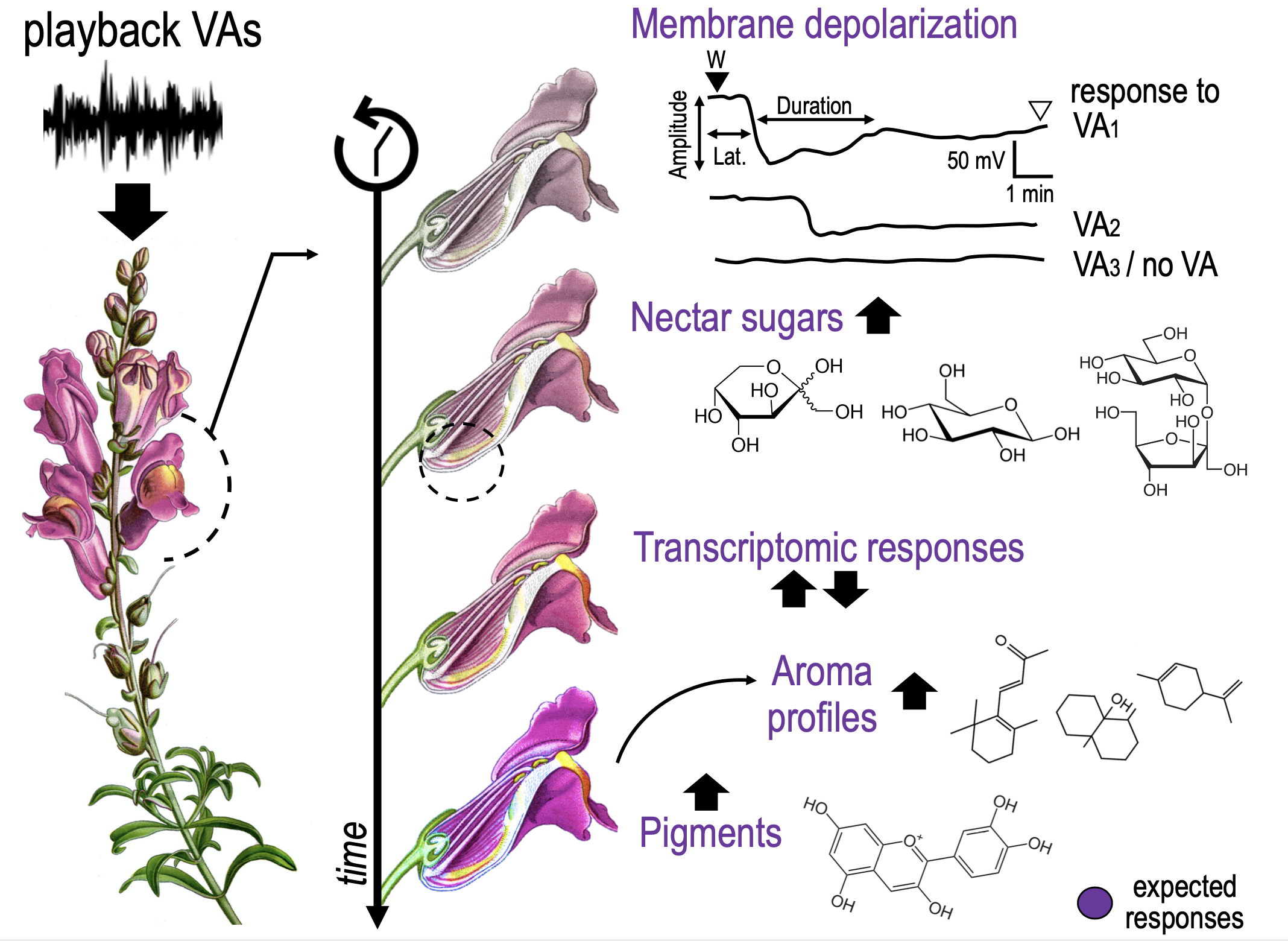
Grapevine is one of the fruit crops of greater relevance in the world. It has been an integral part of human history since its domestication and possesses one of the highest cultivated surfaces both for the production of table fruit and wines. As a consequence, research development in areas regarding grape physiology and metabolism is a main requirement for resource optimization and increased quality production. Likewise, we should face the effects of climate variations, such as the rise of atmospheric CO2, less water availability and the increase of terrestrial radiation and earth surface temperature, all of which have a negative impact on the quality of grapes and wine. Additionally, boundaries for grape cultivation are already shifting and the ozone layer depletion will increase the surface level of UV-B radiation.
Despite grapevine performance is negatively affected in climate change-driven scenarios, studying phenylpropanoid metabolism in grapes and understanding its modulation by the environment represents a challenge to optimize plant resources and to increase quality production. In fact, changes in berry phenylpropanoid composition, positively influenced by increased radiation levels, improve the quality of grapes and wines subsequently produced. Detailed, genome-enabled understanding of gene networks can be translated into tools and resources for the development of crops that respond efficiently to local and changing conditions. In the last years we have been studying the grapevine’s transcriptome responses to environmental factors and agricultural practices related to radiation and temperature (Figure 3.1), including the role of sunlight depletion on anthocyanin and flavonol synthesis in plants grown in commercial vineyards (Matus et al., 2009; Czemmel et al., 2017), and the effects of ultraviolet radiation filtering and irradiating experiments in flavonoid accumulation in mature vines grown in greenhouse and field conditions (Loyola et al., 2016; Czemmel et al., 2017; Matus et al., 2017). Very recently, we further exploited the use of gene networks to understand the role of newly identified transcription factors in the response to environmental stresses such as low temperatures, a detriment condition related to certain wine producing regions such as China (Sun et al., 2018).

Figure 3.1. Experimental setups in commercial vineyards to study the effect pf sunlight depletion of UV-B radition filtering in the berry metabolic composition.For more details see Matus et al., 2009; Loyola et al., 2016; Czemmel et al., 2017; Sunitha et al., in press.
Studying the effects of sunlight depletion and UV-B radiation in the grape’s metabolic program has helped understanding these species’ adaptive measures to environmental fluctuations and represent a source of knowledge for modeling grapevine’s responses to climate change. We have shown that in the field, flavonoid-related genes respond rapidly to changes in light quality, as shown by treatments in which shaded fruits were exposed to light in the late stages of ripening (Matus et al., 2009). In this study it was possible to observe that leaf removal at the onset of ripening had an early diminishing effect on sugar transport and flavonoid synthesis (especially on flavonol accumulation) during the ripening of the berry skin. This work exemplified how the flavonoid content and the genes controlling their synthesis were affected and could be manipulated by viticultural practices related to canopy management.
A second approximation to understand the grapevine’s responses to the environment included the effect of UV-B radiation in the synthesis of flavonoids. In Loyola et al., (2016), we performed irradiation experiments on fruits of 15 year-old potted plants growing in a phytotron under controlled environmental conditions and tested the expression of several genes of the phenylpropanoid pathway and many transcription factors. The bZIP genes HY5 and HYH showed an induction in response to low and high fluence UV-B in correlation to the synthesis of flavonols. A breakthrough derived from this study was to demonstrate that, even though UV-B perception and signaling orchestrates the accumulation of flavonols in almost all terrestrial plant species, the long-term UV-B adaptive mechanisms that grapevines possess are highly efficient in part due to the activation of HY5 and HYH in response to high UV-B, in addition to their conserved photomorphogenic response to low levels of radiation (Figure 3.2). This is in contrast to what occurs in Arabidopsis, where different types of UV-B levels independently activate specific or non-specific signaling pathways. We further explored the capacity of networks to provide a wide overview of gene function and expression behavior of UV-B response genes in grapevine. In Loyola et al. (2016) we determined a high confidence set of HY5 and HYH targets by combining microarray and RNA-Seq data with genome-wide promoter inspections. Despite sharing a large set of common co-expressed genes, we found different hierarchies for HY5 and HYH depending on the organ and stress condition, reflecting both cooperative and partially redundant roles. As a significant breakthrough of this work we identified new candidate UV-B gene markers and demonstrated the robustness of combining promoter and gene co-expression studies as networks were empirically validated.
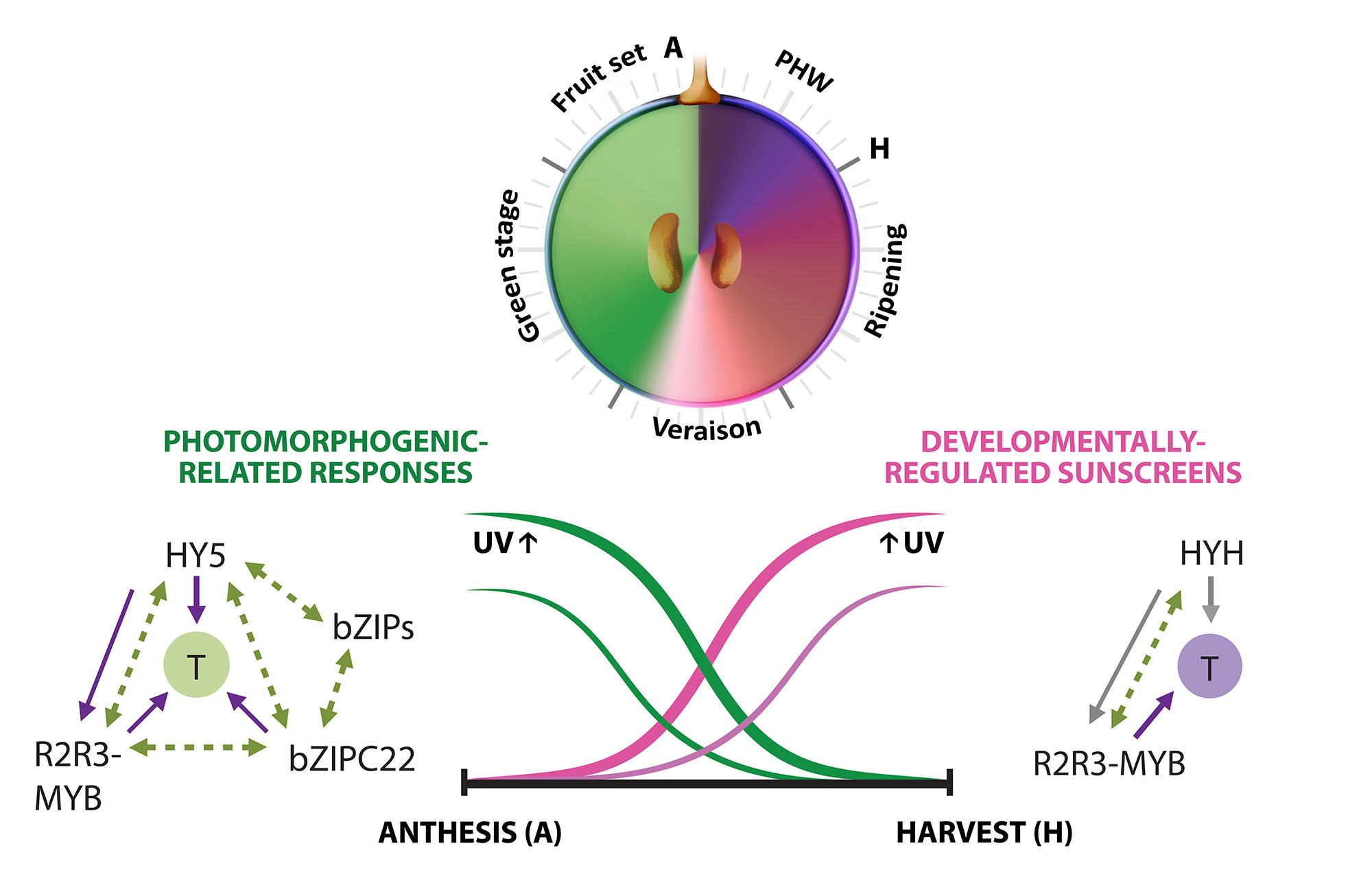
Figure 3.2. Orchestration of UV responses by bZIP transcription factors. The complementary expression patterns of HY5 and HYH in development and their induction by both low and high UV intensities is hypothesized to be in part responsible for the successful adaptation of this species to radiation. Adapted from Matus, 2016. Frontiers in Plant Science.
We have also conducted UV-B filtering experiments in field conditions to test the capacity of UV-B to modulate different steps of the flavonol biosynthetic branch, including glycosylation and sulfourylation modifications. We identified new MYBF1 targets of the flavonol pathway and demonstrated that light and UV-B were able to modify their expression in response to changes in sunlight perception at field (Czemmel et al., 2017). Very recently we have used these same experimental setups to test the transcriptomic changes of small interfering RNAs (miRNAs and TASI-RNAs), some of which show potential effects on flavonoid synthesis (Sunitha et al., in press).
Grape’s growth and productivity can also be greatly affected by other conditions such as low temperatures. For instance, in northern China, all grown vines need to be buried in soil to protect them from extreme winter conditions. This procedure drastically increases the costs of production and decreases the survival rate of vines. Very recently, we have further exploited the use of gene networks to understand the role of newly identified transcription factors in the response to environmental stresses such as cold. In Sun et al., (2018) we constructed an integrated network of co-expression, overexpression RNA-seq data and TF binding landscapes, showing the close relation of the MYB-like TF AQUILO (AcQUIred tolerance to LOw temperatures; after the Greek god of northern winds and winter) with the synthesis of raffinose oligosaccharides (RFOs), a class of osmoprotectants accumulated in response to cold stress. Here we demonstrated the trueness of the network by detecting an increase in RFOs in cold treated 35S:AQUILO overexpressing plants.
In the forthcoming years, we would like to address different bioinformatics-coupled to experimental data to model grape stress responses to multiple factors affected by climate change.
Funding
![]() Funding source: Ministerio de Ciencia e Innovación (Call 2024)
Funding source: Ministerio de Ciencia e Innovación (Call 2024)
Amount (Euros): 212.500 € + PhD contract (FPI)
Period (year- year): 2025-2028
Project title or topic: METACELL. Multi-Omics Meta-Analyses to Elucidate Plant Specialized Metabolic Pathways and Identify Regulatory Factors Using Cell Suspension Cultures as Model Systems. PID2024-163039NB-I00
Role in the project: PI

Funding source: Interreg Next Med
Amount (euros): 2,402,878.27 €
Period (year- year): 2025-2028
Project title or topic: Design of biofortified silk as a nutraceutical bioactive coating for seed and food protection (NutraSHIELD).
Role in the project: Coordinator
State of the project: Grant Contract not signed by University of Valencia due to ‘political issues’.
 Funding source: SUBVENCIONES PROGRAMA PROMETEO PARA GRUPOS DE INVESTIGACIÓN DE EXCELENCIA. Conselleria de Innovación, Universidades, Ciencia y Sociedad Digital. Generalitat Valenciana. (Call 2024)
Funding source: SUBVENCIONES PROGRAMA PROMETEO PARA GRUPOS DE INVESTIGACIÓN DE EXCELENCIA. Conselleria de Innovación, Universidades, Ciencia y Sociedad Digital. Generalitat Valenciana. (Call 2024)
Amount (Euros): 600.000€
Period (year- year): 2025-2029
Project title or topic: DECIPher: Decoding Environmental Communication and Integration Pathways to Enhance Plant Acclimation to Stress
Role in the project: Team Member (Work Package 1 Leader). Project Coordinator: Purificación Lisón; Jaime Martínez

Funding source: Convocatòria d’ajuts per a la recerca en biogenoma (genòmica de la biodiversitat) Institut d’Estudis Catalans (IEC)- Iniciativa catalana per a l’Earth Biogenome Project (CBP). (Call 3)
Amount (Euros): 60.379€
Period (year- year): 2023-2024
Project title or topic: Genomic Insights into Three Locally Adapted Diatoms from the Albufera Coasta Lagoon. IEC-BG4-2024-9
Role in the project: PI-Coordinator (David Navarro-Payá as Co-PI). Team: Ana Conesa, Jerónimo Xirivella, Guillem Salazar.

Funding source: Convocatòria d’ajuts per a la recerca en biogenoma (genòmica de la biodiversitat) Institut d’Estudis Catalans (IEC)- Iniciativa catalana per a l’Earth Biogenome Project (CBP). (Call 2)
Amount (Euros): 99.720€
Period (year- year): 2023-2024
Project title or topic: A taxo-genomic survey of Mediterranean biodiversity and its ecological interactions. IEC-BG2-2023-1
Role in the project: Team Member.
Coordinator: Aureliano Bombarely

Funding source: Convocatòria d’ajuts per a la recerca en biogenoma (genòmica de la biodiversitat) Institut d’Estudis Catalans (IEC). Iniciativa catalana per a l’Earth Biogenome. (Call 2)
Amount (Euros): 12.460€
Period (year- year): 2023-2024
Project title or topic: Development and implementation of standardised processes (pipelines) for genome assembly and annotation. IEC-BG1-2023-4
Role in the project: PI.
![]()
Funding source: Cooperation in Science and Technology Office (COST)
Amount (Euros): 125.000€
Period (year- year): 2022-2023
Project title or topic: The Grapevine Genomics Encyclopedia: an innovative portal to integrate knowledge, resources and services for the grape scientific community and industry. IG17111
Role in the project: Chair
![]()
Funding source: Ministerio de Ciencia e Innovación (Call 2018)
Amount (Euros): 229.900€
Period (year- year): 2022-2025
Project title or topic: VALINET. Validation and visualization of gene regulatory networks of plant specialized metabolism using integrative omics methods. PID2021-128865NB-I00
Role in the project: PI
![]()
Funding source: Human Frontier Science Program (Call2021)
Amount (Dolars): 1.140.000 USD
Period (year- year): 2022-2025
Project title or topic: Good Vibes: how do plants recognise and respond to pollinator vibroacoustic signals? RGP0003/2022
Role in the project: Co-PI
Project Website: https://goodvibes.tomsbiolab.com
![]()
Funding source: SUBVENCIONES PROGRAMA PROMETEO PARA GRUPOS DE INVESTIGACIÓN DE EXCELENCIA. Conselleria de Innovación, Universidades, Ciencia y Sociedad Digital. Generalitat Valenciana. (Call 2020)
Amount (Euros): 597.345€
Period (year- year): 2021-2024
Project title or topic: ENIGmA: Exploring Novel plant-to-plant Intercommunication pathways Governing environmental Acclimation.
Role in the project: Team Member (Unit no.3 Leader).
Project Website: https://enigma.webs.upv.es/
![]()
Funding source: Ministerio de Ciencia e Innovación (Call 2018)
Amount (Euros): 127.050€ + PhD contract (FPI)
Period (year- year): 2019-2021
Project title or topic: NETFRUIT. Systems biology approaches to understand the role of MYB transcription factors in the regulatory networks of secondary metabolism of fleshy fruits. PGC2018-099449-A-I00
Role in the project: PI
Funding source: Ministerio de Ciencia e Innovación (Call 2017)
Amount (Euros): 40.000€
Period (year- year): 2019-2024
Project title or topic: Ramon y Cajal Track: Aproximaciones multi-omicas para caracterizar redes regulatorias implicadas en el desarrollo de frutos carnosos y en vias del metabolismo secundario
Role in the project: PI
Participation as collaborator in International Projects
 Funding source: Núcleo Milenio (ANID, Chile).
Funding source: Núcleo Milenio (ANID, Chile).
Amount (euros): €
Period (year- year): 2025-2028
Project title or topic: Millenium Nucleus in Data Science for Plant Resilience (Phytolearning)
PIs project: José Miguel Álvarez (UNAB), Elena Vidal (Uni Mayor).
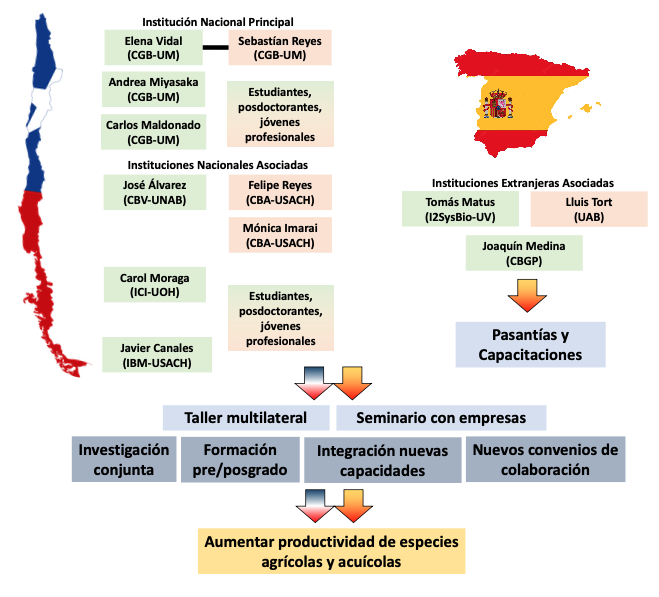 Funding source: FOVI (ANID, Chile): FOMENTO A LA VINCULACIÓN INTERNACIONAL PARA INSTITUCIONES DE INVESTIGACIÓN CONVOCATORIA 2023
Funding source: FOVI (ANID, Chile): FOMENTO A LA VINCULACIÓN INTERNACIONAL PARA INSTITUCIONES DE INVESTIGACIÓN CONVOCATORIA 2023
Amount (euros):
Period (year- year): 2023-2024
Project title or topic: ResilomicsNet: Establecimiento de una alianza internacional para potenciar la resiliencia climática de especies de interés productivo
PI project: Elena Vidal (Uni Mayor).
State of the project: Grant Contract not signed by University of Valencia due to ‘political issues’.
Outreach
Title: Cooking Science Outreach
Funding source: Fundación Española de Ciència y Tecnologia (FECYT) FCT-20-15552
Period (year- year): 2022
Project title or topic: PLAN ANUAL DE ACTIVIDADES DE LA UCC+I DE LA UNIVERSITAT DE VALÈNCIA. Plan de Acción 9: Cocinando la divulgación científica.
Responsible entity: Unidad de Cultura Científica y de la Innovación de la Universitat de València (UCC+i UV).

Period (year- year): 18th May, Yearly-based
Project title or topic: Día de la Fascinación por las plantas
Description:
Activities 2022: Visit from L’IES Vicent Andrés Estellés. Press note & Photo Gallery.

Period (year- year): 2019-2020
Project title or topic: The Secret Life of a Plant Biologist
Funding & Organization: FECYT-MICIN, Fundación Española para la Ciencia y la Tecnología – Ministerio de Ciencia e Innovación & Severo Ochoa-AEI, Programa Centro de Excelencia Severo Ochoa – Agencia Estatal de Investigación. Supported by CRAG (Centre for Research in Agricultural Genomics).